Computational design of spiropyran-merocyanine nonlinear optical photoswitches: from isolated molecules to model aggregates
Marilù G. Maraldi1,2, Angela Dellai1, Frédéric Castet1
1 Institut des Sciences Moleculaires (ISM), Université de Bordeaux, Talence (France)
2 Molecular Chemistry, Materials and Catalysis Division (MOST), Institute of Condensed Matter and Nanosciences (IMCN), Université Catholique de Louvain, Louvain-la-Neuve (Belgium).
Contact: marilu.maraldi@uclouvain.be
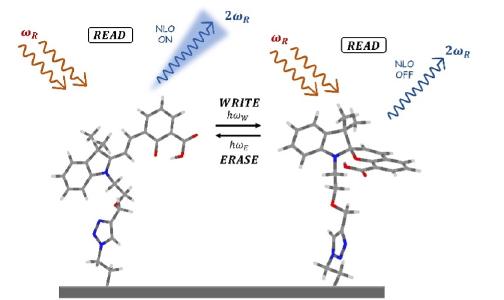
- F. Castet, V. Rodriguez, J.-L. Pozzo, L. Ducasse, A. Plaquet, and B. Champagne, Accounts of Chemical Research 2013, 46, 11, 2656–2665
- E. Orgiu and P. Samori, Advanced Materials 2014, 26, 12, 1827–1845
- B. J. Coe, Chem. Eur. J. 1999, 5, 9, 2464-2471
- S. Casalini, C. A. Bortolotti, F. Leonardi, and F. Biscarini Chem. Soc. Rev. 2017, 46, 40-71
- L. Kortekaas and W. R. Browne, Chemical Society Reviews 2019, 48, 3406–3424
- A. M. Teale, et al. “Dft exchange: Sharing perspectives on the workhorse of quantum chemistry and materials science,” Phys. Chem. Chem. Phys. 2022, Advanced Article